ソフトウェア開発
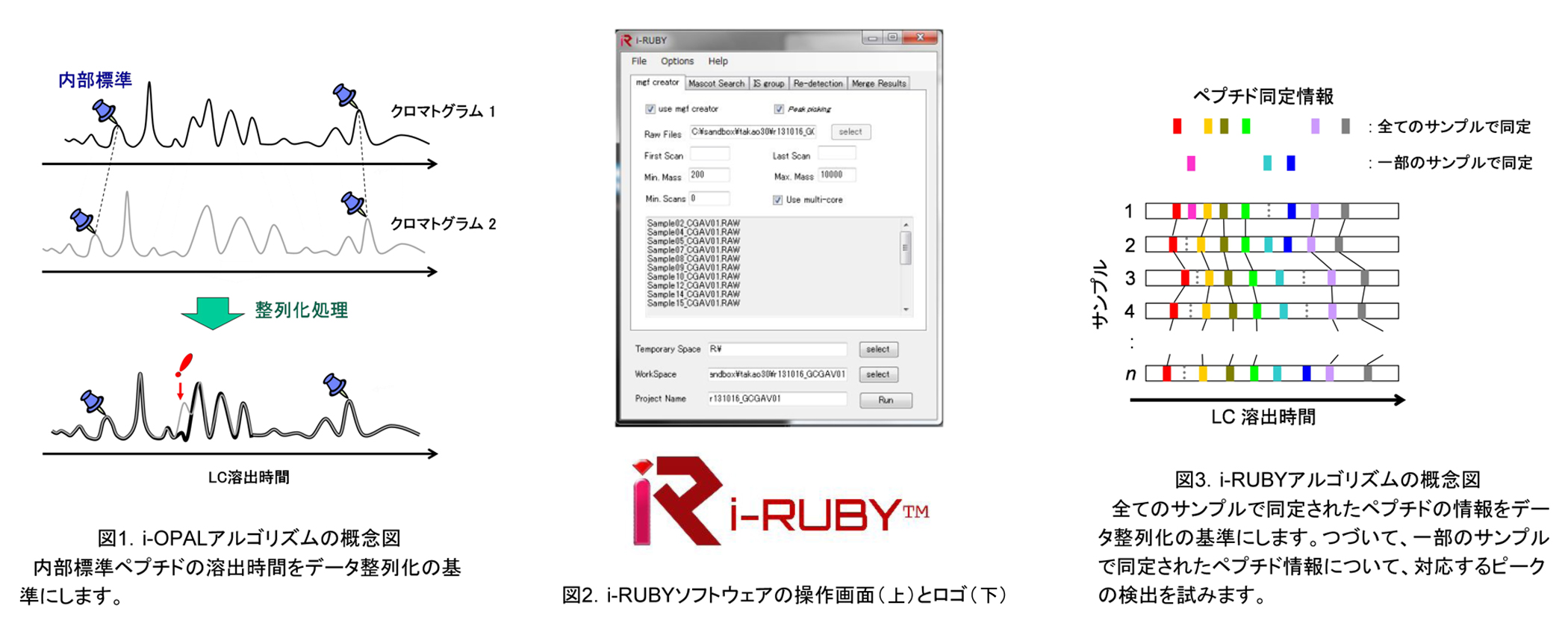
当社では、複数のLC-MS/MSデータを比較するためのソフトウェアを開発しています。開発当初は、測定データ間のデータ整列化に内部標準ペプチドを利用しました(i-OPALアルゴリズム)。最新版(i-RUBY)では、ペプチド同定情報を用いて正確かつ高速のデータ処理を実現しています。
注.現在は、i-OPAL、i-RUBYともに開発と解析サービスを停止しています。
LC-MS/MSデータの整列化処理:i-OPALの開発
i-OPALとは「internal standard-guided Optimal Profile ALignment」の略で、その名の通りデータ整列化処理のために内部標準試料を利用するアルゴリズムです(図1)。当社はこの工夫で特許を取得しました。
プロテオミクスで用いられるLC-MS/MSのLCは、通常1 μL/minを下回る流速に設定されています。この流速ではペプチドの溶出時間を測定ごとに一致させることがたいへん困難です。そこで、イオン検出強度を比較するためには、測定データ間で同じペプチドのイオンシグナルを対応付けることが必要です。この対応付けの処理をシグナルの整列化(alignment)と呼びます。i-OPALアルゴリズムは、内部標準ペプチドの溶出時間を基準にして膨大な検出シグナルデータの整列化を一括でおこないます。
ペプチド同定情報を利用したデータ整列化:i-RUBY
この第2世代のアルゴリズム(図2)では、各ペプチドのイオンシグナルに付随しているペプチド同定情報を利用します。同じ同定情報が付いているシグナルをデータ整列化の基準に用います(図3)。
i-RUBYアルゴリズムの運用には正確なペプチド同定の情報が必須です。このため、Orbitrap質量分析をはじめとする高精度の装置から得られた測定データに適用することができます。
i-RUBYソフトウェアの論文はこちら。