
今さら聞けない プロテオミクスの基礎
プロテオミクスの基礎事項をまとめました。各種教育セミナーなどのために用意した資料も順次公開していく予定です。
目次
・プロテオミクス関連の出来事
・ペプチドを測る
・ショットガン分析
・エレクトロスプレーイオン化法
・生成イオン(プロダクトイオン)スペクトルの例 1
・生成イオン(プロダクトイオン)スペクトルの例 2
・修飾ペプチドの回収: リン酸化ペプチドの場合
・修飾ペプチドの回収: 選択回収の効果
・プロテオミクスで使われている質量分析計
・ボトムアップとトップダウン
・LC-MS/MSデータの二次元表示
・検出強度の再現性
・非標識法と標識法
・同位体原子の導入
・標識法の考え方
・非標識法と標識法の比較
・配列データベース検索の構成要素
・配列データベース検索の考え方
・Target-Decoy検索
・Decoy配列の作り方
・Target-Decoy検索の結果
プロテオミクス関連の出来事
物事の全体を俯瞰するにはまず歴史から、ということで、プロテオミクス関連のおもな出来事を年代順に並べてみました(図)。「Proteomics」の語が初めて現れたのは1994年なのですが、現在使われている基幹技術のいくつかはこの年以前に開発されたので前史も重要です。2000年以降に国際組織の設立や専門誌の創刊が続いています。

プロテオミクス年表
プロテオミクス史の関連では、プロテオミクス以前を含めたおもなタンパク質分析の技術を他サイトで紹介したことがあります。
新技術最前線 新薬開発を目指す人へ【第5回】
プロテオーム分析技術の変遷 すべての分析は自動化に通ず
この寄稿は各技術の自動化に主眼を置きました。併せてご覧ください。
2017年11月14日掲載; 2023年7月10日更新
ペプチドを測る
プロテオミクスの質量分析では、ほとんどの全ての場合でペプチド断片の混合物を直接の測定試料とします。このことを単純な絵で示しました。
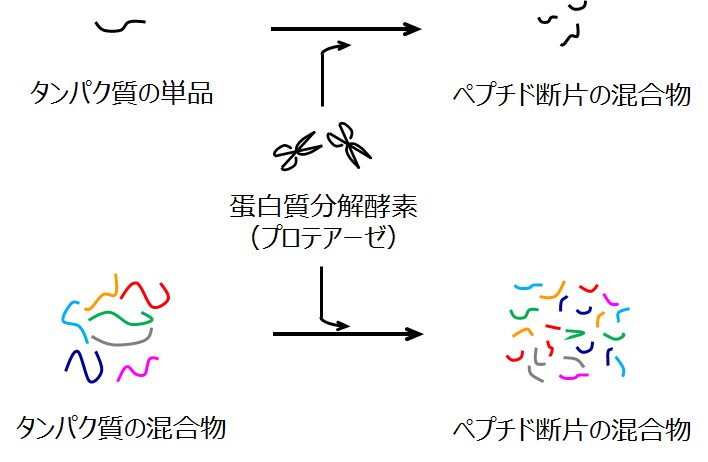
プロテオミクスの質量分析ではペプチドの混合物を測定試料とする
電気泳動でバンドとして展開される単一のタンパク質はもちろんのこと、細胞溶解液のように千種類を優に超えるタンパク質混合物でも基本は同じです。つまり、基質特異性の高いタンパク質分解酵素(おもにトリプシン)を添加して試料タンパク質を短鎖のペプチドにします。こうして得られたペプチド混合物を質量分析計に導入します。プロテオミクス技術のセントラルドグマと言ってよいほど受け入れられている手順であり、この手順を使って現在までに多くの成果が挙がっていることは皆さんもご存知の通りです。
個々のタンパク質分子は、大きさ、電荷、溶媒への溶けやすさなどの性質が実にさまざまです。こういった分子群をそのまま特定の分析システム(この場合はLC-MS/MS)で網羅的に分離検出するのはたいへんですが、タンパク質としてどんな厄介な特性をもっていたとしても、そのアミノ酸配列の中には分析しやすい領域もいくらか含んでいることが期待できます。ペプチドに断片化する理由を少し乱暴に説明するとこんなふうになります。それでも、もともと複雑なタンパク質の組成をさらに面倒なことにしている感は否めません。実際に、ある特定のペプチド同定の情報が複数のタンパク質にまたがって帰属する例や、タンパク質あたりの各修飾体の種類が求めにくくなるなどのため、分析の現場ではデータの解釈に慎重になります。
一方で、タンパク質を分解することなくそのままLC-MS/MSに導入する方法も実用化に向けて研究が進められています。このアプローチは「トップダウンプロテオミクス」と呼ばれていて、質量分析計の性能の向上が大きく貢献しています。上に述べたプロテオミクスの常識も早晩過去のものになってしまうかもしれません。
2018年1月13日掲載
ショットガン分析
続いてペプチドの質量測定です。ペプチド混合物は、微流速の液体クロマトグラフにタンデム質量分析計が接続した分析システム (LC-MS/MS) に導入されます。
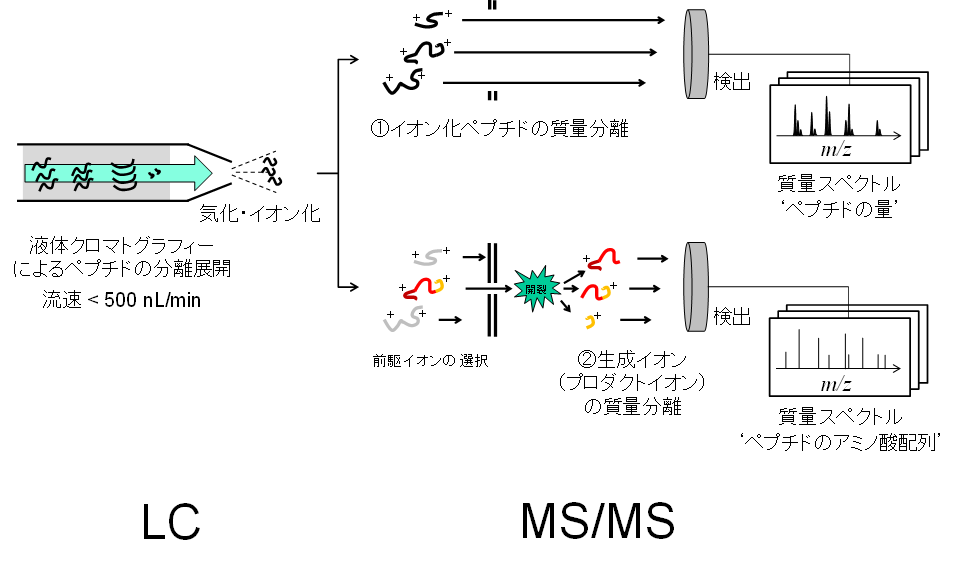
ショットガン分析の模式図
上図ではデータ依存的取得法 (Data-dependent acquisition, DDA) の様子を示しました。質量分析計内では2つの測定モードでそれぞれイオン化ペプチド群の質量スペクトルとプロダクトイオンの質量スペクトルを取得します。測定中は両走査が高速で切り替わり、たとえば前者1回の直後に後者の走査が10回連続で続くような編成を取ります。開裂反応に供するイオン化ペプチドを各イオンの検出強度に応じて(データ依存的に)自動選択するためにこの名 (DDA) があります。なお、イオン化ペプチド単位で選択せずに、設定したm/z幅に収まるイオンをm/zをずらしながら一斉に開裂反応に供する走査モードも開発されています。このデータ非依存的取得法 (Data-independent acquisition, DIA) は、DDAよりもプロテオームの同定網羅性が高い測定法として有力です。
2022年5月11日更新
エレクトロスプレーイオン化法
エレクトロスプレー (Electrospray ionization, ESI) は、LCとMS/MSのインターフェースとしてプロテオミクスに限らず広範に使われているイオン化法です。
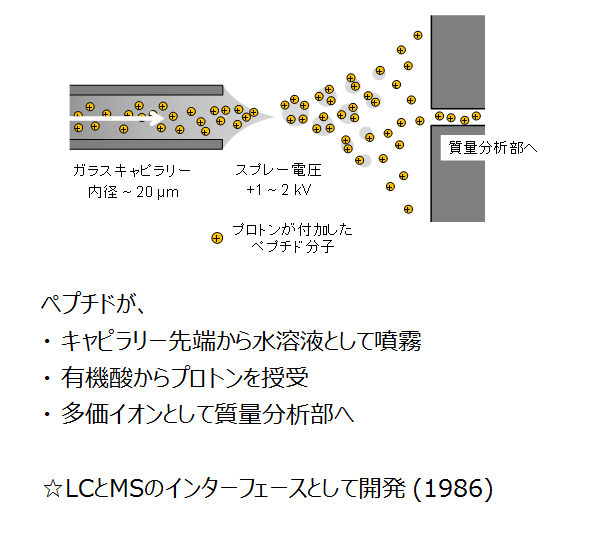
エレクトロスプレーイオン化法 (ESI) の原理
ESIの原理は上の図に示した通りです。LC-MS/MSに搭載した場合は、分析カラムの出口に付けたキャピラリーニードルの先からLCの移動相(液体)がポンプ圧で押し出されます。プロテオミクスにおけるペプチドのLC-MS/MSでは、ほとんどの事例でLCは逆相モードを採り、かつLC移動相中のギ酸がペプチドにプロトンを供与します。
2018年10月23日掲載; 2023年12月19日更新
生成イオン(プロダクトイオン)スペクトルの例 1
質量分析計内に導入されたペプチドのプロトン付加分子は、アルゴンやヘリウムなどの不活性ガスの衝突によって開裂反応を起こします(不活性ガスの代わりに窒素を使う質量分析計もあります)。この反応をごく単純化したポンチ絵を図の下の方に示しました。
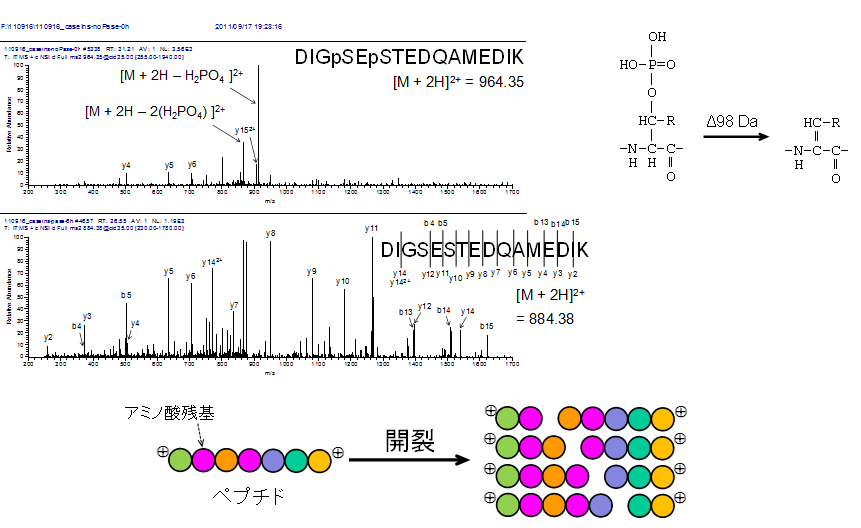
リン酸化ペプチドとその非修飾体のプロダクトイオンスペクトル
開裂反応はおもにペプチド結合で起こりますが、基本的はペプチド分子1個当たりランダムに1か所だけ開裂するので、このように数珠式で描いたような開裂産物の混合物となります。個々の開裂産物のことを生成イオン(プロダクトイオン)と呼びます。
この混合物の質量スペクトル(プロダクトイオンスペクトル)は、理想的にはラダー(はしご)様の形状になります。ある合成ペプチドのプロダクトイオンスペクトルを図の中段に示します。比較的強度の高いピークとピークの間の差がアミノ酸残基の質量に相当している点が重要です。つまり、この質量差を丁寧に拾っていくと当該ペプチドのアミノ酸配列を読み取ることができます。
一方、修飾が付いたペプチドはプロダクトイオンスペクトルの様子が大きく変わることがあります。さきほどのペプチドとアミノ酸配列は同じだけれどもリン酸基が2か所入った修飾体では、プロダクトイオンスペクトルがとたんに貧弱になります(図の一番上)。リン酸基が外れる開裂反応がペプチド結合に優先して起こるためです。
このように、プロダクトイオンスペクトルはペプチドの一次構造の情報を含みます。しかしながら、プロテオミクスでは質量スペクトルからアミノ酸配列を読み取っていく場面は稀でして、多くのプロテオミクス研究はプロダクトイオンスペクトルとアミノ酸配列の両情報を照合するアプローチを採ります。この点は後述します。
2018年10月23日掲載; 2023年12月26日更新
生成イオン(プロダクトイオン)スペクトルの例 2
もう一つだけプロダクトイオンスペクトルの例を挙げます。
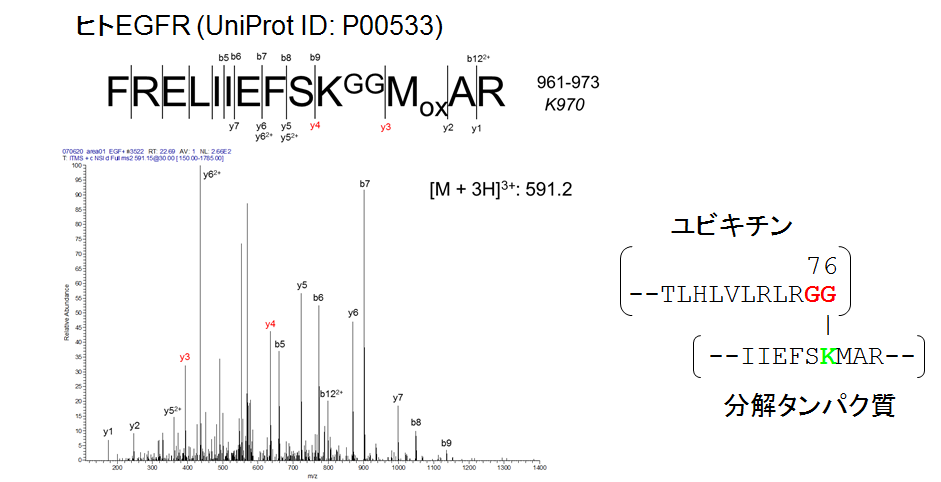
ユビキチンン化ペプチドのプロダクトイオンスペクトル
細胞内発現タンパク質のユビキチン化は、ユビキチンのC末端カルボキシル基が標的タンパク質のリジン残基のアミノ基に結合することで生じます。ユビキチンのC末端配列が「-Arg-Gly-Gly」なので、ユビキチン化タンパク質をトリプシンで分解すると、トリプシン活性の基質特異性にしたがってArgとGlyの
間で加水分解が起こります。その結果、ユビキチン化されたリジン残基がこの図に示すようにGly-Gly修飾体として検出されます。
Gly-Gly修飾リジンを抗原としたモノクローナル抗体が開発されており、トリプシン分解ペプチド断片の混合物からGly-Gly修飾体を選択的に回収する手順が確立しています。この試料調製手順は、ユビキチン化の基質タンパク質を網羅的に同定する目的で汎用されています。
2018年10月23日掲載: 2023年12月26日更新
修飾ペプチドの回収: リン酸化ペプチドの場合
ペプチド断片の質量分析をもとにしたプロテオミクス技術は、タンパク質の翻訳後修飾を網羅的に同定するときにも応用できます。分析を効率よく進めるために、一般には、注目した修飾基が付いているペプチド断片を非修飾ペプチドから選択的に分画します。このアプローチが一番成功している修飾がリン酸化です。図では、チタニア樹脂によるリン酸化ペプチドの回収の原理を示しました。
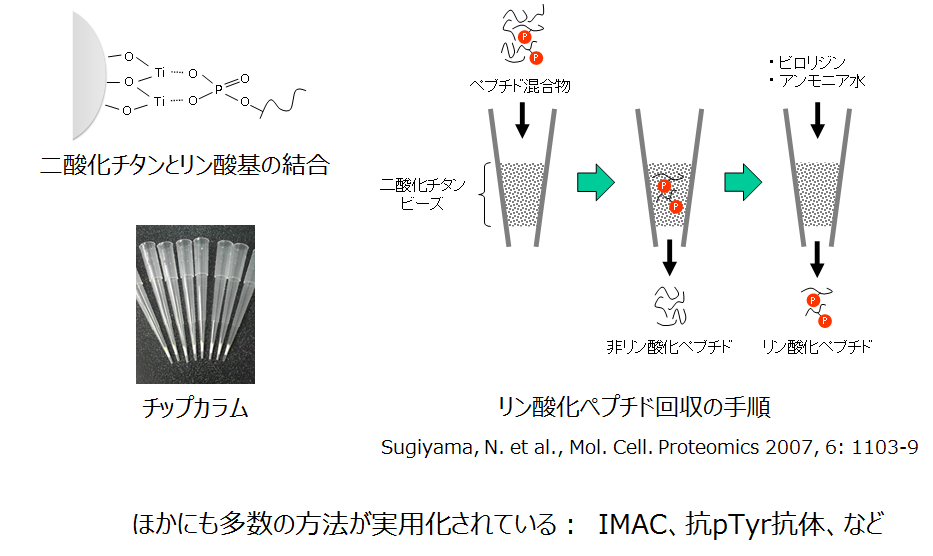
チタニア樹脂によるリン酸化ペプチドの回収
リン酸基と二酸化チタンとの間のイオン結合よってリン酸化ペプチドが保持されます。この親和性は、リン酸化されているアミノ酸残基(おもにセリン、トレオニン、チロシン)の種類と数に依りません。
比較的微量の試料でも処理できるように、写真で示したとおりマイクロピペットのチップの先にチタニア樹脂を詰めておこなうプロトコールが普及しています(詰め済みの市販品もあります)。
また、ここで紹介したチタニア法のほかにも、リン酸化チロシン抗体を用いた手順など多くの方法が発表、開発されています。
2018年10月23日掲載; 2023年12月28日更新
修飾ペプチドの回収: 選択回収の効果
リン酸化ペプチド回収の効果の一例です。前のページで紹介したチタニア法を用いて回収処理前後のペプチド同定情報を比べました。
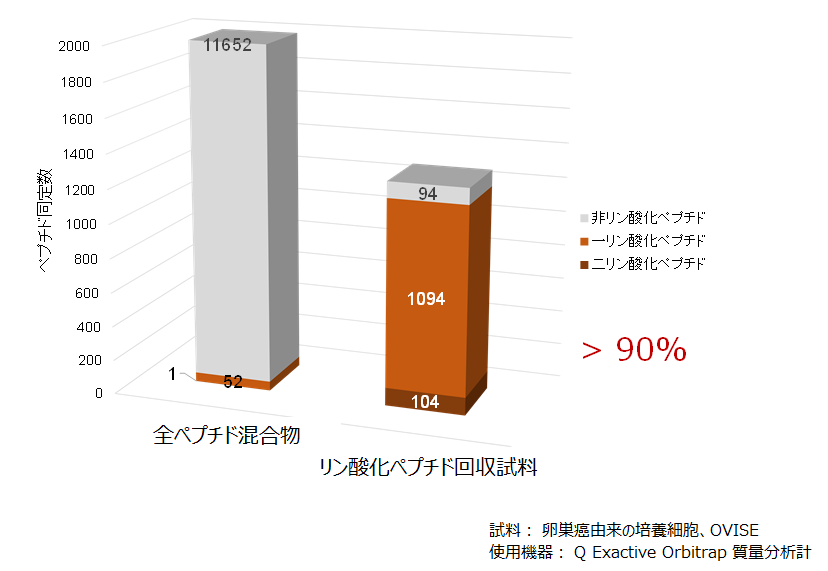
リン酸化ペプチドの回収の効果
培養細胞を出発試料として、タンパク質の抽出変性とトリプシン加水分解を経てペプチド断片の混合物を調製しました。この混合物を直接LC-MS/MS測定すると合計で10000件以上のペプチドが同定されたのですが、リン酸化ペプチドの同定は53件にとどまりました。同じペプチド混合物を前ページのチタニアチップに通した場合は、その吸着画分から同定されたリン酸化ペプチドは1198件に上り、しかも全ペプチド同定に占める割合は90%を超えました。他の培養細胞でもほぼ同様の成績が得られています。
リン酸化ペプチドはリン酸基の高い親水性のため、スプレーイオン化の際の気化の過程で水などの溶媒が離れにくいことが知られています(ある専門家の方が「溶媒離れが悪い」と表現していました)。そこで、非リン酸化ペプチドと一緒に居ると気化過程の競争に負けてしまい、結果としてリン酸化体の検出効率が低下します。この現象を避ける意味でも、リン酸化ペプチドの選択回収は必須の前処理手順です。
2018年10月23日掲載; 2023年12月28日更新
プロテオミクスで使われている質量分析計
質量分析部による分類

現在のプロテオミクスでは様々な型式の質量分析計が使われています。上図では質量分析部に注目して質量分離の原理を3種類に分類しました。たとえば、サーモフィッシャーサイエンティフィック社のOrbitrap質量分析計は、この中のイオントラップ型に含まれます。2つ以上の原理を組み合わせたハイブリッドタイプの機器も上市されているので、実際の分類はこんな単純ではありません。ともあれ、各々の研究の目的に適った質量分析計を選択することが肝要です(分析の網羅性を重視するか、それとも標的分子の定量分析か、など)。
2018年7月10日掲載; 2023年6月26日更新
ボトムアップとトップダウン
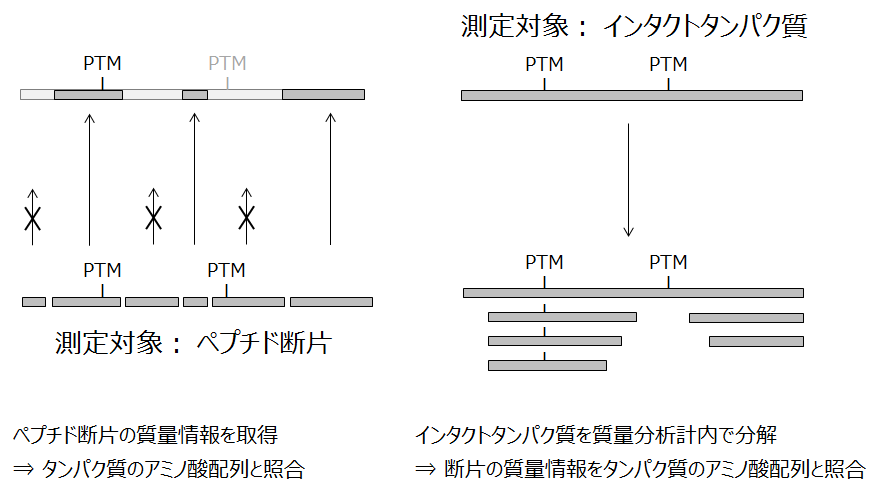
ボトムアッププロテオミクス(左)とトップダウンプロテオミクス(右)
質量分析を基盤とするプロテオミクスには、ペプチド断片ではなくインタクトのタンパク質そのものを測定の対象にすることがあります。このアプローチはトップダウンプロテオミクス (Top-down proteomics) と呼ばれています。逆に、ボトムアッププロテオミクス (Bottom-up proteomics) は、ペプチド断片の混合物のLC-MS/MSとMS/MSデータの配列データベース検索とを組み合わせた手法を指しています。「ショットガンプロテオミクス (Shot-gun proteomics)」とほぼ同じ意味で使われています。
図では両者の分析手順を対比できるように表現しました。
トップダウン法では、インタクトタンパク質を質量分析計内で開裂させ、その結果生じたペプチド断片の質量情報をタンパク質のアミノ酸配列と照合します。インタクト分子の質量も計るので、当該タンパク質分子の全体像を修飾の種類と数も含めて把握することができます。なお、この場合の「インタクト」とは、生体内で発現、機能している構造が共有結合レベルで維持されている状態を指します。
ボトムアップ法では、どうしても見つからない断片が出てきます。このため、「このタンパク質に付いているリン酸基はいくつか?」といった一見簡単な問いにも答えにくいのです。
それでもトップダウン法はボトムアップ法に比べて今のところ適用例がずっと少ないです。質量分析に導入するための、タンパク質の網羅的な分離展開の方法が確立されていませんし、また、さまざまな性質をもつタンパク質分子を一斉に気化、イオン化するのも困難だからです。
トップダウン法とボトムアップ法の折衷、たとえば、インタクトタンパク質を臭化シアンなどで大きめの断片にしてから測定するアプローチも試みられています。これはミドルダウン (Middle-down) 法と呼ばれています。
2018年10月23日掲載; 2024年1月9日更新
LC-MS/MSデータの二次元表示
ショットガン分析(ボトムアップ法)の解説に戻ります。
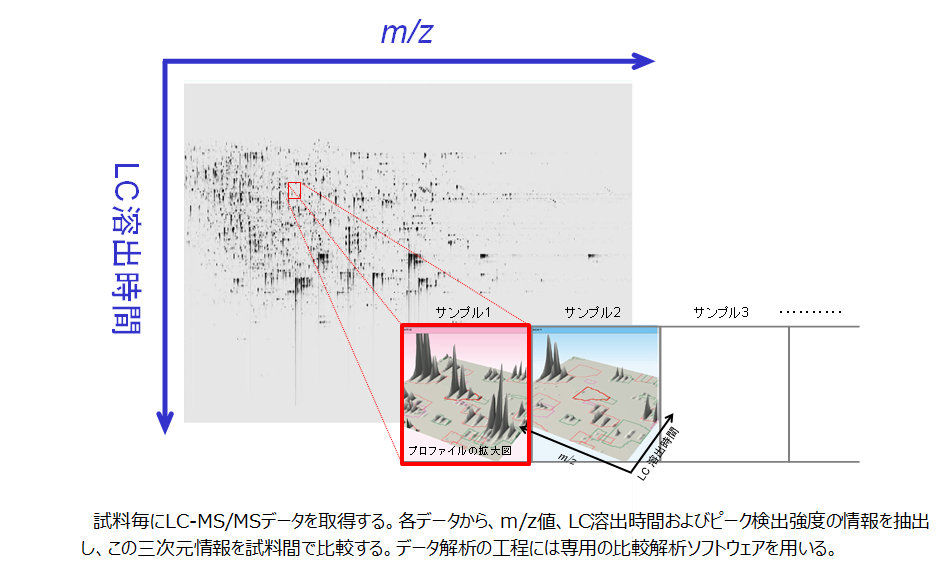
LC-MS/MSデータの二次元表示
ペプチド混合物のLC-MS/MSデータは図のように視覚的に表現することができます。単一のLC-MS/MS測定データを、質量電荷比とLC溶出時間を両軸とした平面として展開すると、各プロトン付加ペプチド分子が平面上で点状に分布します。この表現における検出強度軸は平面の垂線に相当します(三次元目)。
あとで説明する非標識プロテオミクスでは、試料ごとに取得したLC-MS/MSデータを相互に比較します。比較に際しては、LC-MS/MSデータ間で同じ位置にある検出ピーク同士を連結させる情報処理をおこないます(整列化処理と呼びます)。整列化処理を経てはじめて検出強度の正規化処理や試料間比較が可能になります。
2018年10月23日掲載; 2024年1月9日更新
検出強度の再現性
前の図で述べた、非標識プロテオミクスのデータ再現性の一例です。
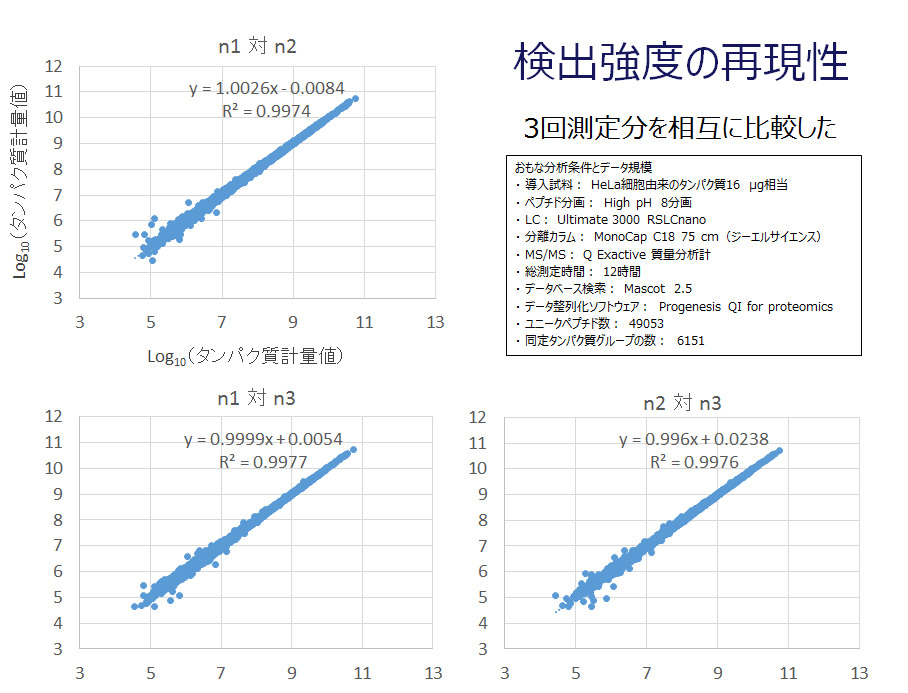
タンパク質計量値の対角線表示
培養細胞から全タンパク質を抽出変性し、トリプシン加水分解を経てペプチド混合物を調製しました。続いてペプチド混合物を8つの画分に分け、各画分を3回ずつLC-MS/MSに供しました。取得したMS/MSデータを測定回ごとに8画分統合したうえで、MS/MSデータ3セットからそれぞれ得られたタンパク質同定計量情報を相互に比較しました。
比較表現の一つとして対角線散布図を示します。この図から、LC-MS/MSデータ間の計量比較が探索レベルの分析試験に十分耐えうると判断しました。
2018年10月23日掲載; 2024年1月9日更新
非標識法と標識法
他のオミクス研究と同様に、現在のプロテオミクスではプロテオーム間の計量比較分析が頻繁に行われています。質量分析を用いた計量比較の手法は非標識法と標識法に大別されます。
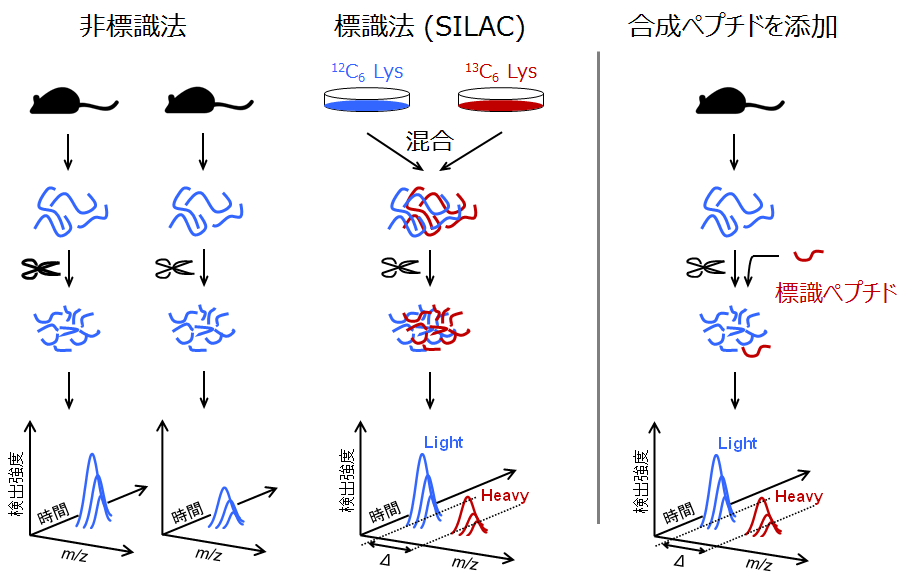
プロテオーム計量比較の工程
プロテオミクスでは標識法の方が先に実用化されました(2000年代の初頭)。標識法の一つであるSILACの工程を図の中央に示します。
SILAC: stable isotope labeling by amino acids in cell culture
分子内の原子を13Cや15Nに置換した必須アミノ酸を培地に溶かして細胞を培養すると、代謝反応によってタンパク質中の当該アミノ酸残基が全て標識体に換わっていきます。この方法は代謝標識と呼ばれています。代謝標識した細胞と標識していない細胞のプロテオームを比べる場合は、両細胞のタンパク質抽出物を混合し、その後は一直線にプロテアーゼ分解とLC-MS/MSに進めます。
一方、非標識法では、各試料から個別にペプチド混合物の測定データを取得します(図の左側)。現在は微流速LC-MS/MSシステムの運用のノウハウが蓄積されるとともに、システムそのものの堅牢性と安定性が高くなっています。このため、異なるLC-MS/MSデータの間でも実用上充分正確に計量的な比較ができるまでになっています。
なお、特定のタンパク質やペプチド断片を定量する場合は、対象とするペプチド断片の安定同位体標識体(標識ペプチド)を試料に既知量添加した上で分析に供します(図の一番右)。この方法では、ペプチド断片とその帰属元のタンパク質の濃度を求めることができます。
2018年10月23日掲載; 2024年1月12日更新
同位体原子の導入
標識法の解説の続きです。
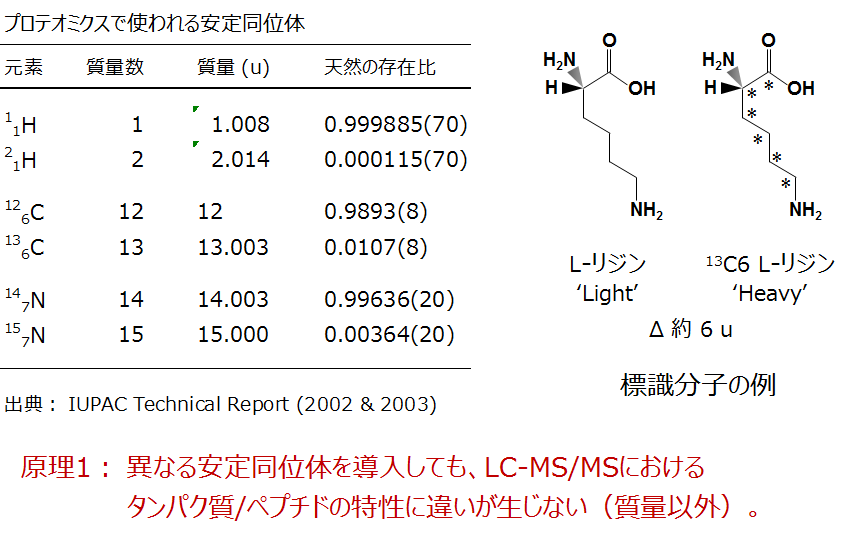
安定同位体原子による標識
プロテオーム計量比較には、おもに炭素または窒素の安定同位体原子が利用されています。水素は、重水素に置き換えると逆相LCにおけるペプチドの溶出時間が変動することがあるので、現在ではほとんど適用例がありません。炭素と窒素では、ペプチドに重い方の原子(13Cと15N)を導入してもLC-MS/MSにおける特性が質量以外は変わらないので、試料間の正確な計量比較が可能になります。
標識体の例としてリジン分子の構造を示します。リジンの炭素6個がすべて13Cに置き換わると、置き換わる前と比べて質量が約6 u(6ダルトン)だけ増加します。この重いリジンを培養液に入れて細胞を継代していくと細胞内タンパク質のリジン残基が重い方に換わっていきます。
細胞内の代謝反応を利用した標識法は、より正確な計量比較が求められる分析実験に利用されています。
2018年10月23日掲載; 2024年1月12日更新
標識法の考え方
標識法では、各試料からそれぞれ生じる標識体(非標識体)の検出強度を、単一のLC-MS/MS測定データの中で比較します。この点が重要です。極端な言い方ですと、標識法ではLC-MS/MS測定の再現性に多くの注意を払う必要がありません。
標識法における最も簡単な比較の様子を示しました(図)。

標識法を用いたプロテオーム計量比較
標識処理を施していない試料から生じたペプチドのプロトン付加分子「Light」と、重い同位体原子で標識されたペプチドに由来する「Heavy」です。両者の質量差は質量電荷比 (m/z) の軸に現れます。質量以外の物理化学特性は同じなので、LC溶出時間は重なります。こうして検出された両ピークの強度を割り算して、当該ペプチドが帰属しているタンパク質の試料間倍数値を求めます。
現在までにさまざまな標識法が開発されてきました。なかでも上述のSILACと、標識化合物をペプチド断片に結合させる方法(iTRAQとTMT)が普及しています。これら以外の標識法が使われているのを見つけるのがたいへんなくらいです。
SILACに代表される代謝標識法は原理的に培養細胞にのみ適用できます。一方でiTRAQとTMTにはその制限がありません。また、iTRAQとTMTではプロダクトイオンスペクトルに含まれる標識化合物由来のピークを計量比較の対象とします。
2018年10月23日掲載; 2024年1月12日更新
非標識法と標識法の比較
この表では、両者の長所と短所を筆者なりに整理してみました。たいへん大雑把な評価です。標識法を一括りにしている点もお許しください。
表.非標識法と標識法の比較
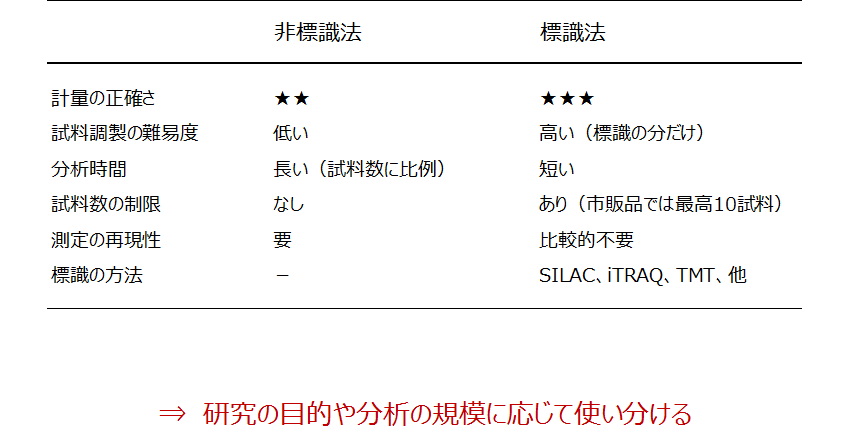
計量の正確さ、すなわち試料間の倍数値の精度は一般に標識法の方が高いです。
試料調製の手順は、標識法では標識処理の分だけ煩雑になります。一方、非標識の場合は、LC-MS/MSに試料を導入するまで多試料を一様に調製する必要があります。そこで、調製の各工程を厳密に制御する目的で試料調製の自動化が課題になります。
分析時間(LC-MS/MS測定の総時間)は非標識法の方が長いです。比較に供する試料を1件ずつの連続測定に供します。したがって、LC-MS/MSシステムの長期間にわたる測定安定性と堅牢性が非標識プロテオミクスの肝になります。
非標識法には計量比較に供する試料の数に制限がありませんが、実際は解析ソフトウェアとその搭載PCの性能に依存します。標識法では標識重原子の数が試料数の上限を決めます。最近では最大16試料を一括で比較が可能な標識試薬が市販されています。
測定の再現性は、上述のとおり非標識法における必須の要件です。
以上をまとめますと:
非標識法はバイオマーカー探索をはじめとした多試料のスクリーニングに適しています。
標識法は、細胞内リン酸化のキネティクスを追うような、正確な計量情報を必要とする研究に向いています。
どちらの方法が絶対的に優れているわけではなく、分析の目的や規模によって使い分けるのが最善です。
2018年10月23日掲載; 2024年1月12日更新
配列データベースの構成要素
アミノ酸配列データベース検索(以下、「配列DB検索」)は、現在のプロテオミクスでもっとも頻繁に用いられているデータ解析手法です。配列DB検索に必要な要素を3点挙げます。

・タンデム質量分析 (MS/MS) データ: プロテオミクスではペプチド断片の測定データの一つです。MS/MSによって当該ペプチド断片のプロダクトイオンスペクトルを取得しますが、MS/MSデータはプロダクトイオンスペクトルだけでなく、前駆イオンのm/z値や価数の情報も含んだ呼称です。
・配列データベース検索ソフトウェア: 単に検索エンジンとも言います。MS/MSデータとアミノ酸配列情報を照合し両者の同一性の確率を数値化する機能を有します。マトリックスサイエンス社のMascotやサーモフィッシャーサイエンティフィック社のSEQUESTが古くから使われています。
・アミノ酸配列データベース(配列DB): タンパク質毎に列挙されたアミノ酸配列の情報です。UniProtなどの公共サイトから、ヒトやマウスなどの生物種単位でも自由に出力できます。
各要素を組み合わせた検索の原理はこの下の図を使って説明します。
2018年10月23日掲載; 2023年6月26日更新
配列データベース検索の考え方
配列DB検索の原理を線対称型の図で表現しました。
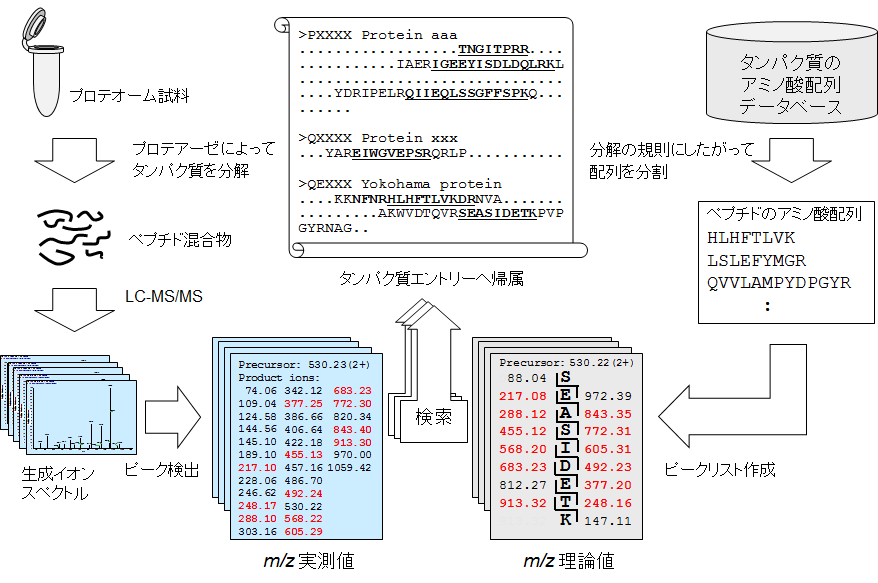
配列データベース検索によるペプチド/タンパク質の同定
図の左側はおもに「物質の流れ」です。分析対象試料から抽出・調製したタンパク質(群)を、特定のアミノ酸残基に作用するタンパク質分解酵素(プロテアーゼ)によってペプチド断片に分解します。続いて、ペプチド断片の混合物をLC-MS/MSに供し、個々のぺプチドからMS/MSデータを取得します。このあとは検索エンジンのピーク検出機能を使って、MS/MSデータ中のプロダクトイオンのm/zを数値情報として出力します(m/zの実測値)。
図の右側は情報処理です。配列DB内のタンパク質のアミノ酸配列情報を、試料調製に用いたプロテアーゼと同じ規則で分割します。分割してできたアミノ酸配列からはどんなm/z値のプロダクトイオンが出てくるか予想できます(m/zの理論値)。m/z理論値と上記の実測値との間で照合の度合いを数値化したうえで、各照合結果をもとのタンパク質に帰属していきます。以上のとおり、検索の中身を大雑把に追ってみました。
検索結果はタンパク質またはペプチド断片の同定一覧として出力します。分析の目的によって両者を使い分けます。例えば、ゲルバンドの同定などではタンパク質同定一覧の出力のみで足りることが多いです。リン酸化プロテオミクスのような翻訳後修飾の分析では、ペプチド同定の一覧表に記載されている情報を用いて各種データ解析をおこないます。
配列DB検索は、MS/MSデータに合うアミノ酸配列を配列DBから選択する手法です。したがって、検索に適用した配列DBに含まれていないアミノ酸配列はそもそも同定しようがありません(例外あり)。同定の取りこぼしを避けるためには、配列DBの選択と構成がたいへん重要です。たとえば、ヒト由来の培養細胞から網羅的なタンパク質同定一覧を取得したいときには、当然ながらヒト遺伝子約20,000件を検索の対象とします。試料の由来が複数の生物種にわたる場合は配列DBの方でも同じ組み合わせにするなど工夫します。ただし、配列DBの規模を必要以上に大きくすると偽同定(次項参照)の確率が上がってくるのでご注意ください。
もう一つの注意点として、同定結果、すなわちMS/MSデータとアミノ酸配列との照合の信頼性を挙げます。上図に示した通り、ペプチド断片の単位で各タンパク質を同定します。このとき同定一覧には必ずと言っていいほど間違いが含まれます(「偽同定」や「偽陽性ヒット」と言います)。間違う理由はMS/MSデータの貧弱さをはじめとしていくつかあるのですが、現在のプロテオミクスでは統計的な処理をしつつ一定程度の間違いを許容する方針が一般的です(後述)。
2018年7月10日掲載; 2023年6月27日更新
Target-Decoy検索
配列DB検索の結果、最初にペプチド断片の同定一覧表が得られます。同定一覧には、同定されたペプチド断片のアミノ酸配列と、各ペプチド同定の信頼度を表す数値が含まれます。

Target-Decoy検索の考え方
同定の信頼度の頻度分布を模式的に表しました(図の左上)。横軸に信頼度(この図では「同定スコア」と呼びます)を置き、縦軸に当該ペプチド同定の数をプロットすると、十分な数の同定情報が得られている場合はこのように2つの極大値をもつ分布になります。
真の同定、すなわち正解のペプチド同定の山と偽ヒットの山は完全には分かれません。そのため、偽陽性ヒットを除こうとして同定スコアの高いところで線引きをすると、多くの真ヒットを失います。逆に、同定スコアの閾値を低く設定した場合は、同定一覧のなかに含まれる偽陽性ヒットがそれだけ増えることになります。このあたりの事情は、健康診断のがんマーカー検査などとよく似ています。なお、図では真のヒットと偽のヒットを色別に示しましたが、現実には真偽はわかりません(正解は神のみぞ知る、です)。
ともあれ、プロテオミクスでは偽陽性ヒットを一定の割合で含むように同定スコア値を情報処理によって求めます。前置きが長くなりましたが、この情報処理のことを「Target-Decoy検索」と呼びます。
Target-Decoy検索では、検索の対象である通常の配列DB(Target配列DB)のほかに、Target配列DBから新たに作った囮の (Decoy) 配列DBを用意します。Decoy配列DBは現実にはあり得ないアミノ酸配列を集積したものでして、その作り方は次のページをご参照ください。
実際の検索工程では、MS/MSデータをTarget 配列DBとDecoy配列DBに別個に照合します。Decoy配列(ありえない配列)との照合によって得られたペプチド同定はすべて間違いだとみなします。
これらの偽ヒットのスコア分布は、Target 配列DBを用いた検索で得られた偽陽性ヒットに近似します。このようにして見立てた偽陽性ヒットの分布をもとにして、偽陽性ヒットの割合 (False discovery rate, FDR) が一定の値になるように同定スコアの閾値を調整します(図の左下)。プロテオミクス研究ではFDRを0.01または0.05に置くことが多いです。
2018年10月23日掲載; 2024年1月13日更新
Decoy配列の作り方
Decoy配列はもとの配列 (Target) 配列を反転させて作ります。反転処理を模式的に表しました(下図)。

反転配列の作り方の一例
反転配列 (Reversed sequence) はペプチド断片のアミノ酸組成が反転させる前との間で違いがありません。長さも同じです。また、反転の規則を決めておけばTatget配列DBからDecoy配列DBが1種類だけ出来上がります。
反転処理の他にアミノ酸配列をランダムに並べ替えるやり方もあります。筆者が試した限りでは、反転配列とランダム配列ではTarget-Decoy検索の結果に大きな違いがみられませんでした。
2018年10月23日掲載; 2024年1月13日更新
Target-Decoy検索の結果
Target-Decoy検索の実施例です。全ペプチド混合物とリン酸化ペプチド試料からそれぞれ得られた検索結果を比べました。
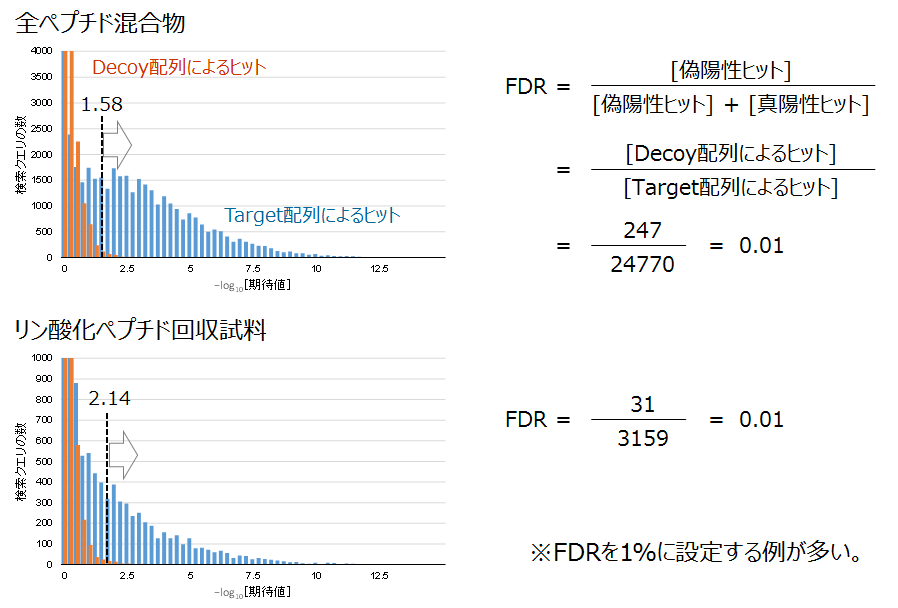
ペプチド同定の信頼度の分布
ひとつめの棒グラフ(上)は、培養細胞から調製したペプチド断片の混合物を分析した結果のスコア分布です。ふたつめのグラフ(下)は、同じペプチド断片試料をリン酸化ペプチドの回収処理に供した上で取得したMS/MSデータがもとになっています。両グラフにはFDRを1%に設定したときのスコア閾値を点線で示しました。
ペプチド同定の信頼度の分布に合わせてスコアの閾値を設定する様子がつかめると存じます。また、リン酸化ペプチドの回収試料では真陽性ヒットの山がはっきりしていなくて、スコアの閾値も高くなりました。この一因として、リン酸化ペプチドのプロダクトイオンスペクトルが非リン酸化体に比べて貧弱になる傾向を挙げます。「生成イオン(プロダクトイオン)スペクトルの例 1」のページもご参照ください。
2018年10月23日掲載; 2024年1月13日更新










